This web page was produced as an assignment for Genetics 564, an undergraduate course at UW-Madison
What is phylogeny?
Phylogeny is the relationship between organismal lineages as they evolve through time [2]. Organisms can be considered to be connected based on the passage of genes throughout evolutionary history, and these connections can be used to create trees representing similarity between organisms. Phylogenetic trees can be considered an effort to retrace evolutionary history through mathematical models that determine similarity between characteristics shared between organisms [3].
To generate phylogenetic trees, programs such as Clustal W2, MUSCLE, or T-COFFEE will align amino acid sequences of proteins and identify regions of similarity and divergence. After a multiple sequence alignment, programs will use one of two techniques for measuring the closeness between sequences in the alignment. Percent identity calculates the percentage identity of sequences at each aligned position. BLOSUM creates a score matrix that assigns each amino acid in a sequence a score according to its frequency. Additionally, programs can use methods to refine tree structure. For instance, the neighbor joining method of programs is employed to create trees with the shortest branch lengths. In contrast, the average distance methods constructs trees that group similar sequences in to each node, and branch length reflects the shared sequence between branches within the node.
To generate phylogenetic trees, programs such as Clustal W2, MUSCLE, or T-COFFEE will align amino acid sequences of proteins and identify regions of similarity and divergence. After a multiple sequence alignment, programs will use one of two techniques for measuring the closeness between sequences in the alignment. Percent identity calculates the percentage identity of sequences at each aligned position. BLOSUM creates a score matrix that assigns each amino acid in a sequence a score according to its frequency. Additionally, programs can use methods to refine tree structure. For instance, the neighbor joining method of programs is employed to create trees with the shortest branch lengths. In contrast, the average distance methods constructs trees that group similar sequences in to each node, and branch length reflects the shared sequence between branches within the node.
Neighbor Joining tree using percent identity
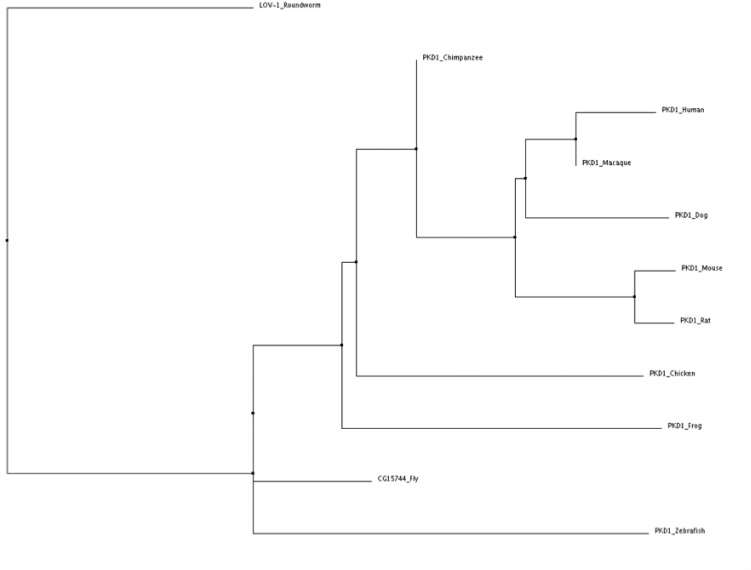
Tree obtained using Clustal W2. Tree verified using MUSCLE.
Average distance using BLOSUM62
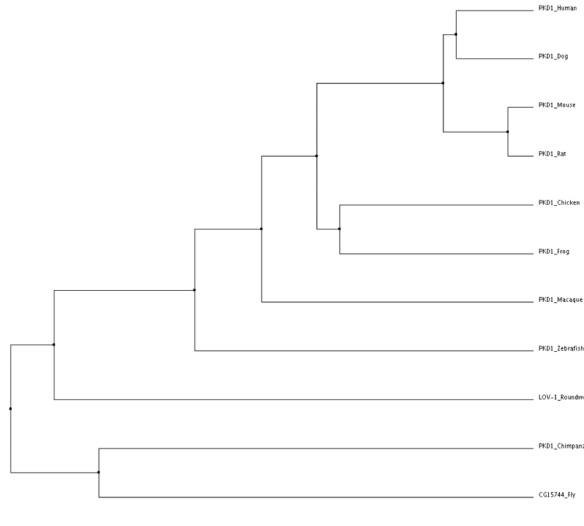
Tree obtained using Clustal W2 and verified using MUSCLE
Average Distance Tree using percent identity
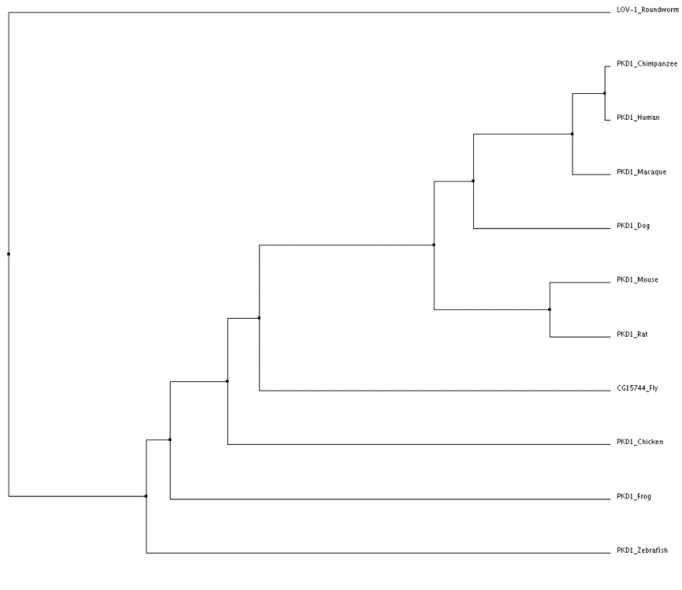
Tree obtained using Clustal W2 and verified using MUSCLE
Neighbor joining tree using BLOSUM62
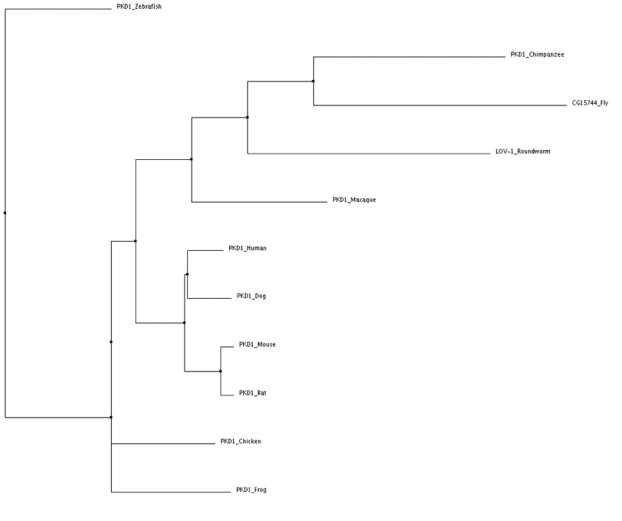
Tree identified using Clustal W2 and verified using MUSCLE
Analysis
These phylogeny trees yielded unexpected results. Organisms that are often considered to be evolutionarily closely related (such as humans and chimpanzees) are not always grouped in to the same node. Additionally, the trees also often yielded phylogenetic trees with great discrepancies between each other. This could be because of the differences in domain structure in polycystin-1 between organisms. Polycystin-1 is a large protein with many exons and domains -- closely related organisms might simply have a different overall domain composition but similar functional domain composition. Alternately, these differences could be due to splicing differences between organisms. More specific analysis of these differences in domain structure or splicing might yield insight in to the functional significance of each part of the particularly large polycystin-1 protein.
References
|
Site created by: Elizabeth Roeske Last updated: 5.14.2014 University of Wisconsin-Madison: Genetics 564 |
